BioMaS Galaxy¶
A modular pipeline for Bioinformatic analysis of Metagenomic AmpliconS (Galaxy Version 0.1.0)
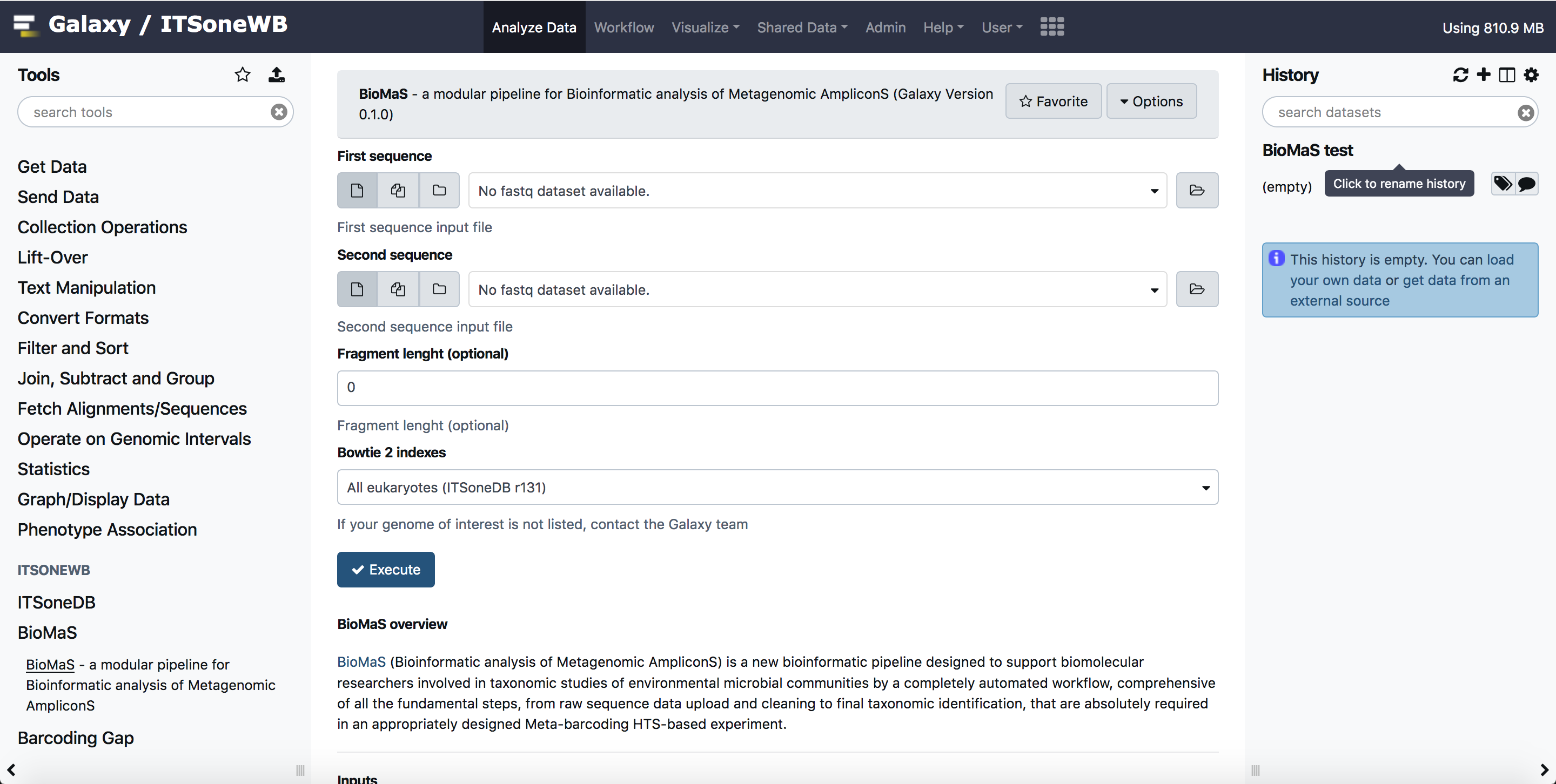
BioMaS Galaxy usage¶
Select your input files:
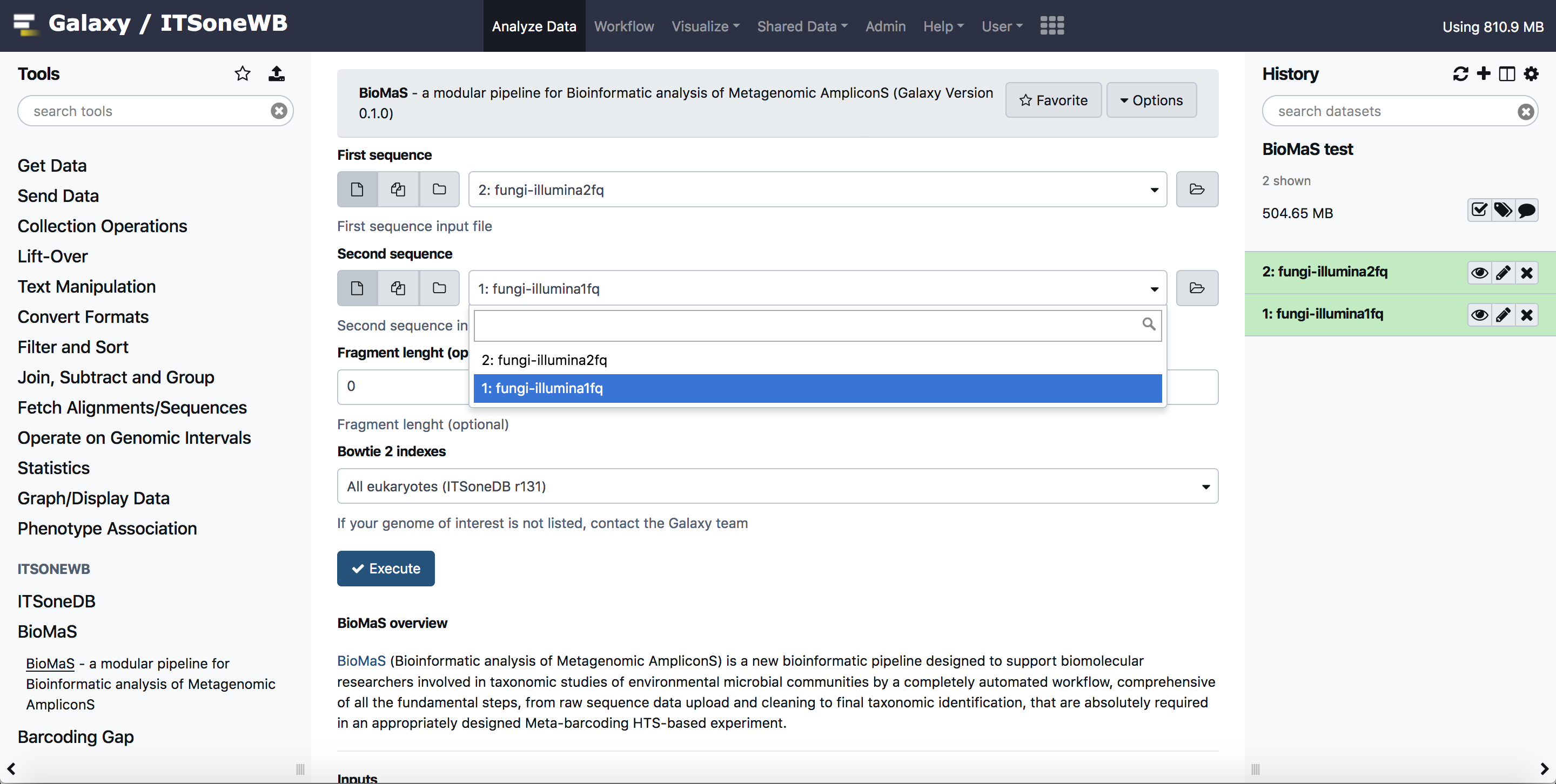
Submit your job:
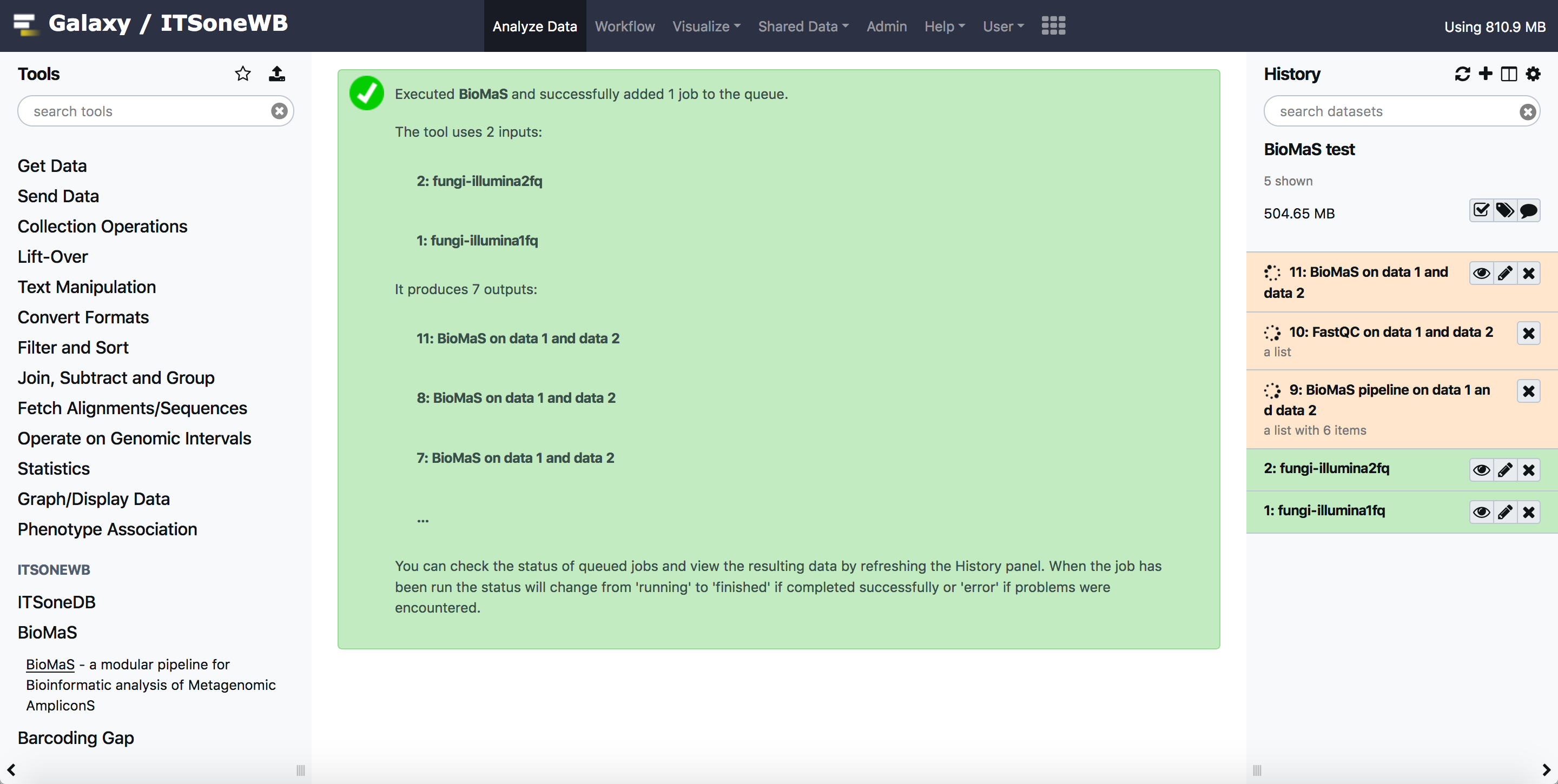
BioMaS Output
The output ov BioMaS tools is composed by two output collection: the fastqc output and the BioMaS pipeline output
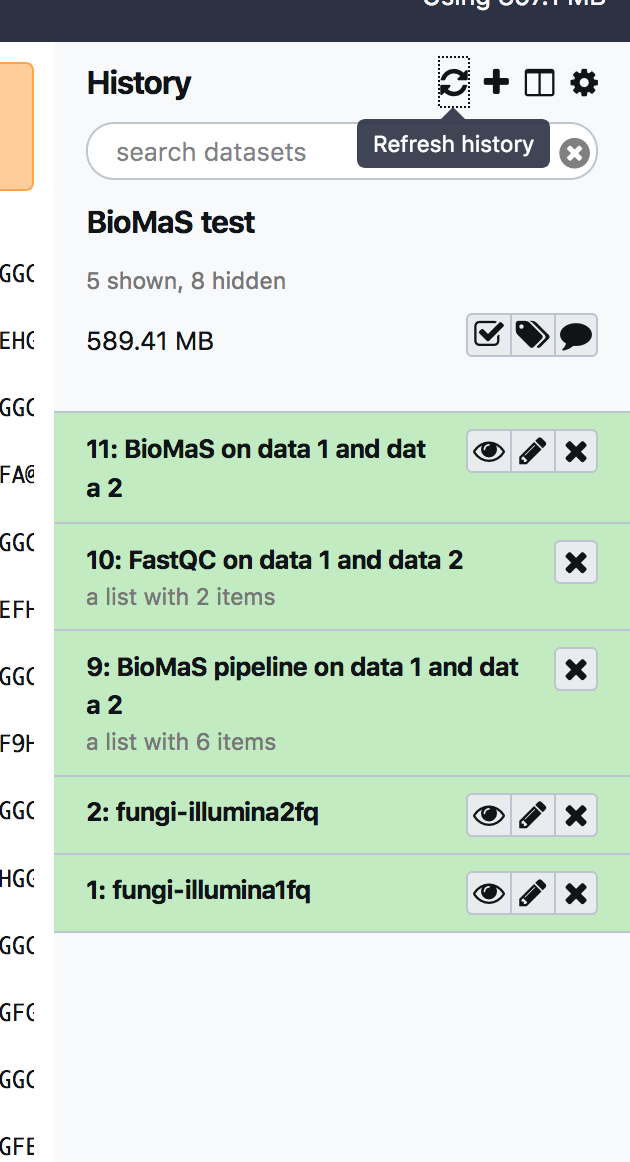
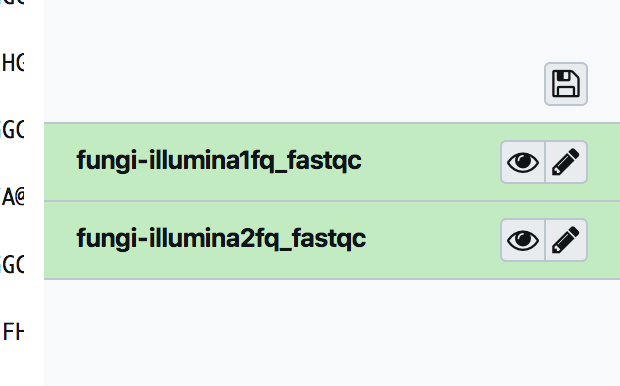
by clicking on the “eye” icon is possible to preview the results, while expanding each entry is possible to download the results.
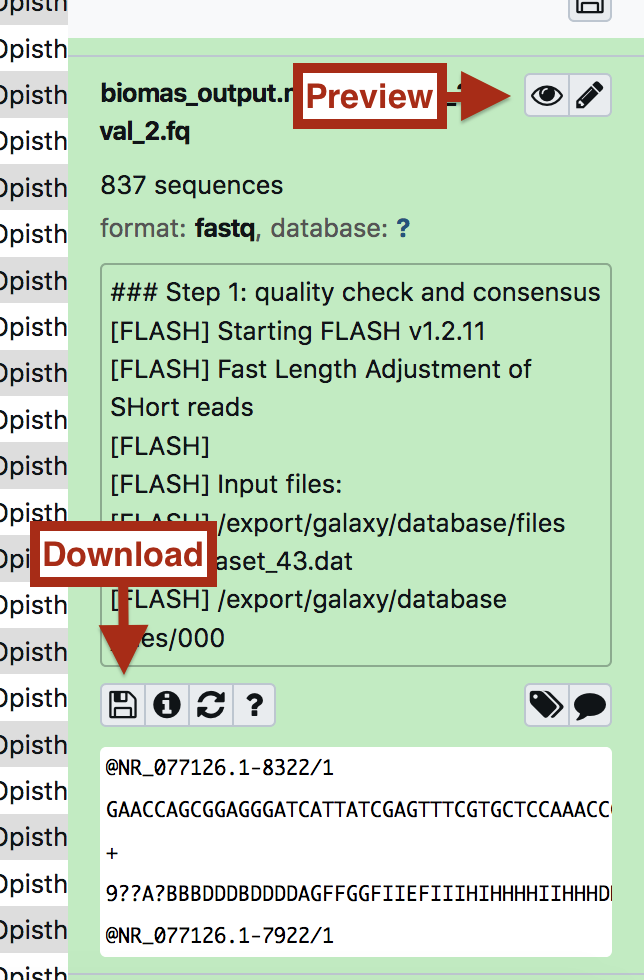
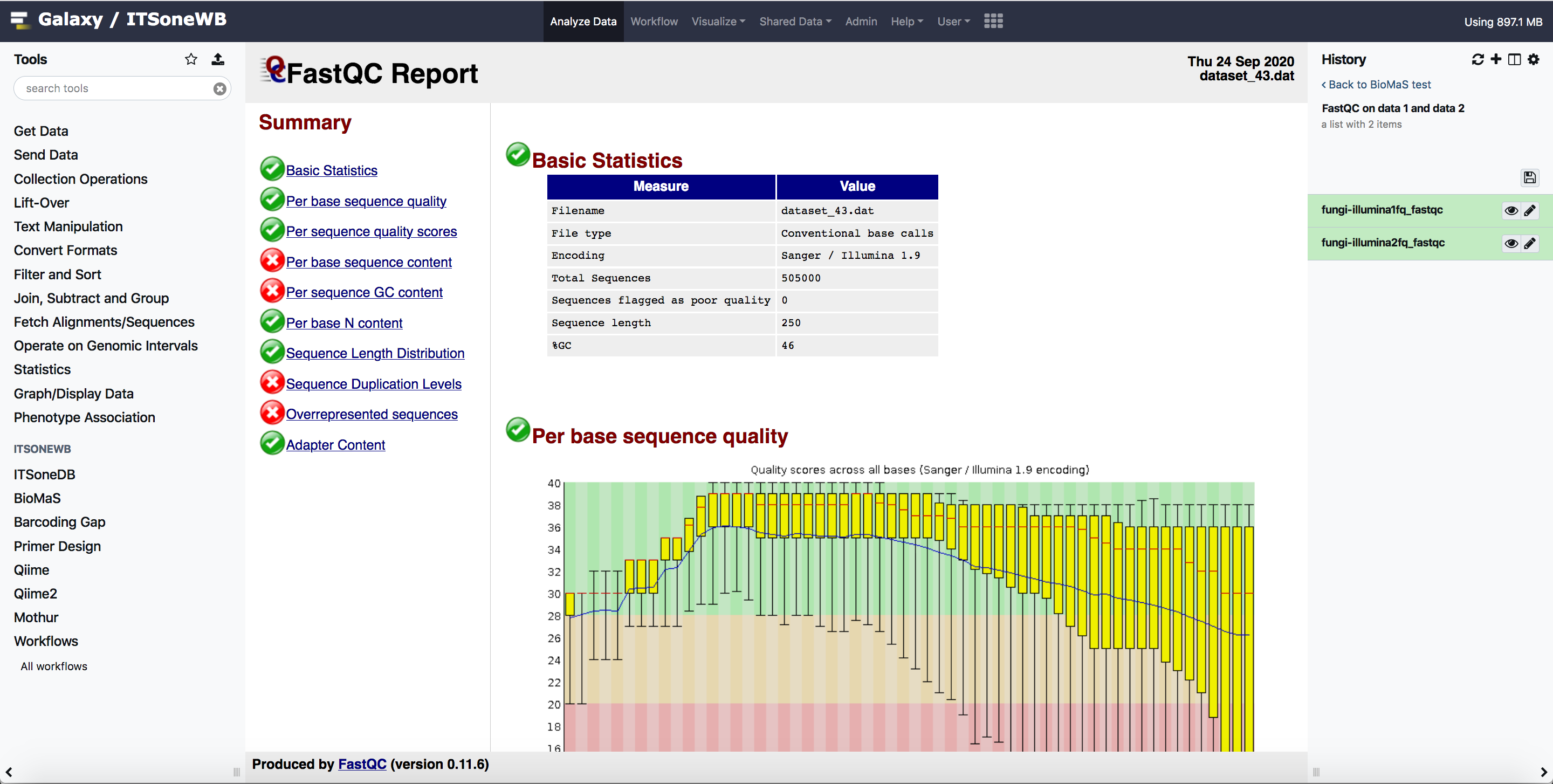
The fastqc output can be easily shown in galaxy:
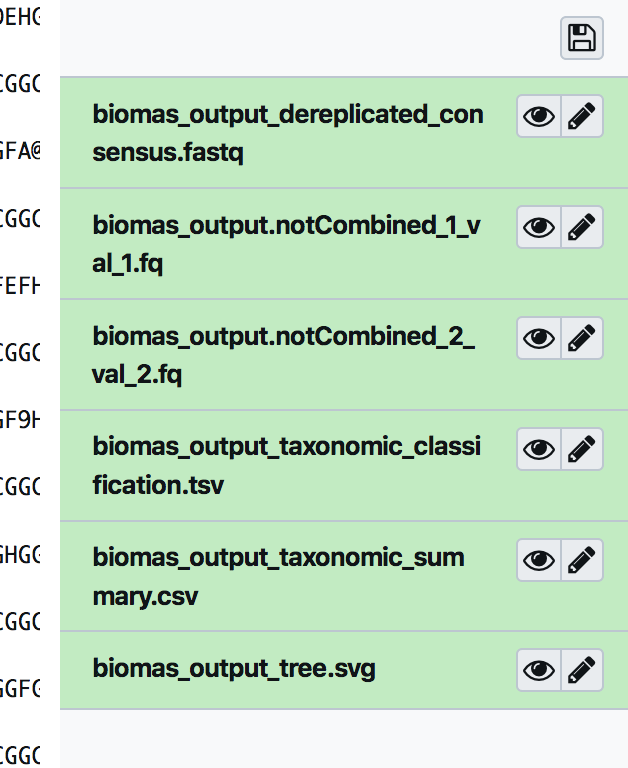
The biomas output is made by:
the dereplicated output;
the not combined output;
the taxonomc classification;
the taxonomic summary;
the SVG Tree (you have to download it to display).

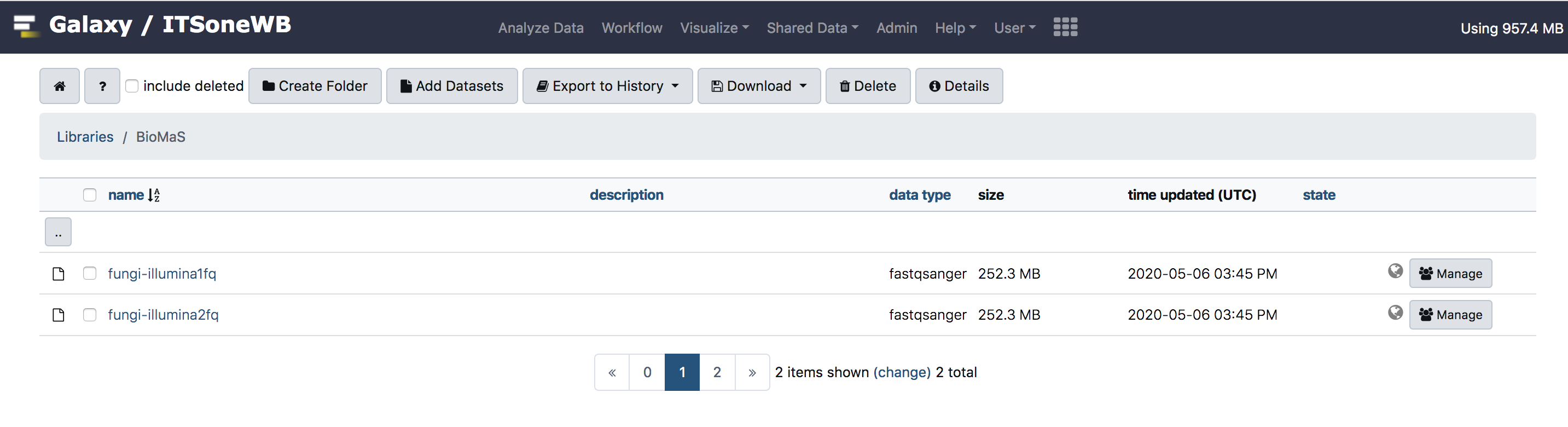
Testing BioMaS Galaxy¶
For testing purpose, we uploaded test input for BioMaS, in Galaxy shared library. These can be exported to Galaxy history and used also by anonymous users.