ITSoneDB vs. shotgun mapping tool¶
The ITS1 shotgun mapping service allows to identify and eventually taxonomically classify ITS1 regions in metagenomic shotgun data.
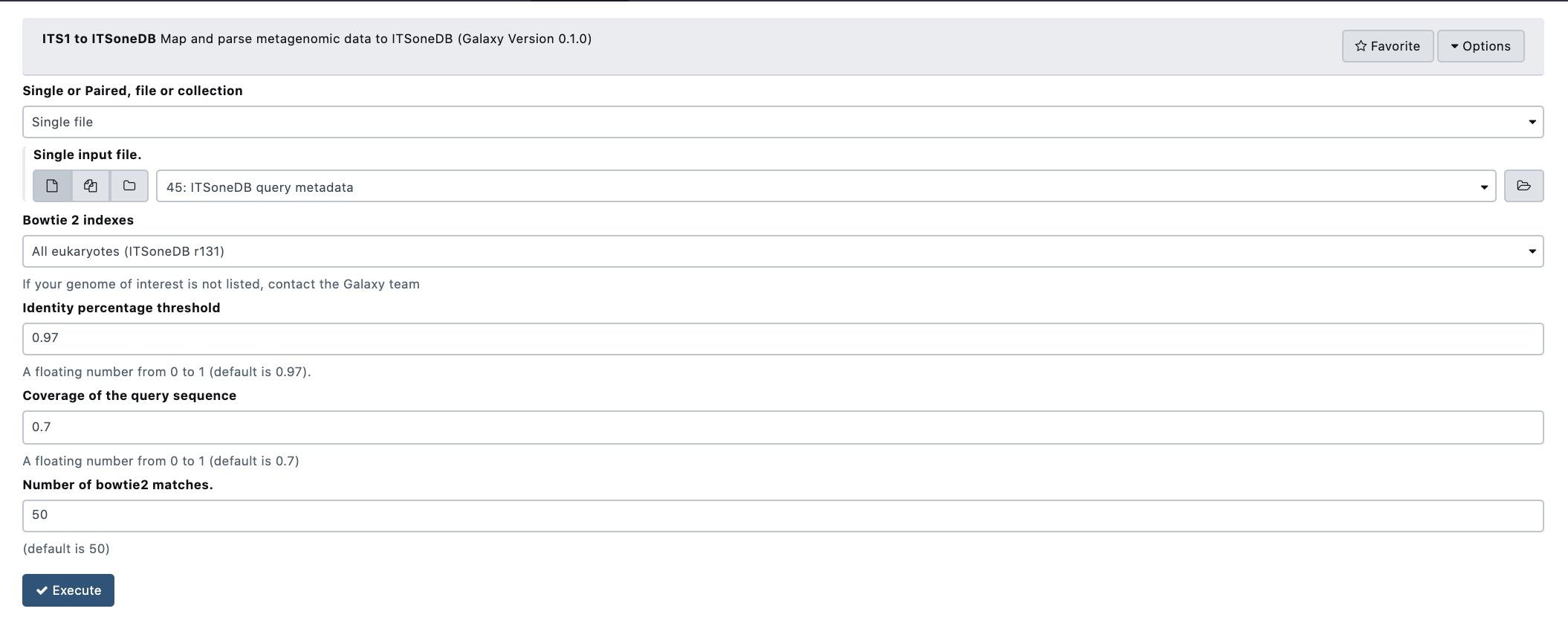
Supplementary Figure 9: A snapshot of the tool setup mask.
ITSoneDB vs. shotgun mapping tool is able to analyse both single-end (SE) and paired-end (PE) fastq files. It exploits query sequence mapping on the ITSoneDB collection by using bowtie2, in both end-to-end (similar to a global alignment) and local modality. The resulting alignments are then filtered according to query coverage and similarity. Finally, a list of ITS1 sequences are returned.
It is important to note the choice between the single-end and the paired-end mapping may influence the results. In particular, in PE mapping both the reads must pass the query coverage threshold.

Supplementary Figure 10: A schematic representation of the mapping schema implemented in the tool. Red and blue lines correspond, respectively, to paired- and single-end reads. For PE reads, R1 and R2 represents the forward and the reverse reads, respectively.
In Figure upplementary Figure 10 4 hypothetical mapping situations are represented. In the first one (A) both the PE mates align to the ITS1 reference for more than 70% of their length, so assuming the identity percentage (measured as the mean of the similarity obtained by each mate) is higher than the imposed threshold, this alignment is retained. In the second case (B), the R1 aligns to the reference for less than 70% and consequently the PE alignment is completely filtered-out. Single-end cases are more simple because we check the requirements for each read independently (C is retained and D not).
Taking into account the mapping and filtering modalities, the user needs to choice between the following parameters:
Single or Paired, file or collection: type of input files;
Bowtie 2 indexes: ITSoneDB reference collection;
Identity percentage threshold: the identity percentage filtering threshold to consider the alignment relevant (default ≥ 97%);
Coverage of the query sequence: the query coverage filtering threshold to consider the alignment relevant (default ≥ 70%);
Number of bowtie2 matches: maximum number of relevant alignments retrieved per each query sequence (or paired-end sequence).
In Supplementary Figure 10, a simulation of single-end data analysis by using default parameters is shown.
The result of the analysis is a tubular text file listing the query sequences matching with ITS1 sequences.
Galaxy Usage¶
Map and parse metagenomic data to ITSoneDB.
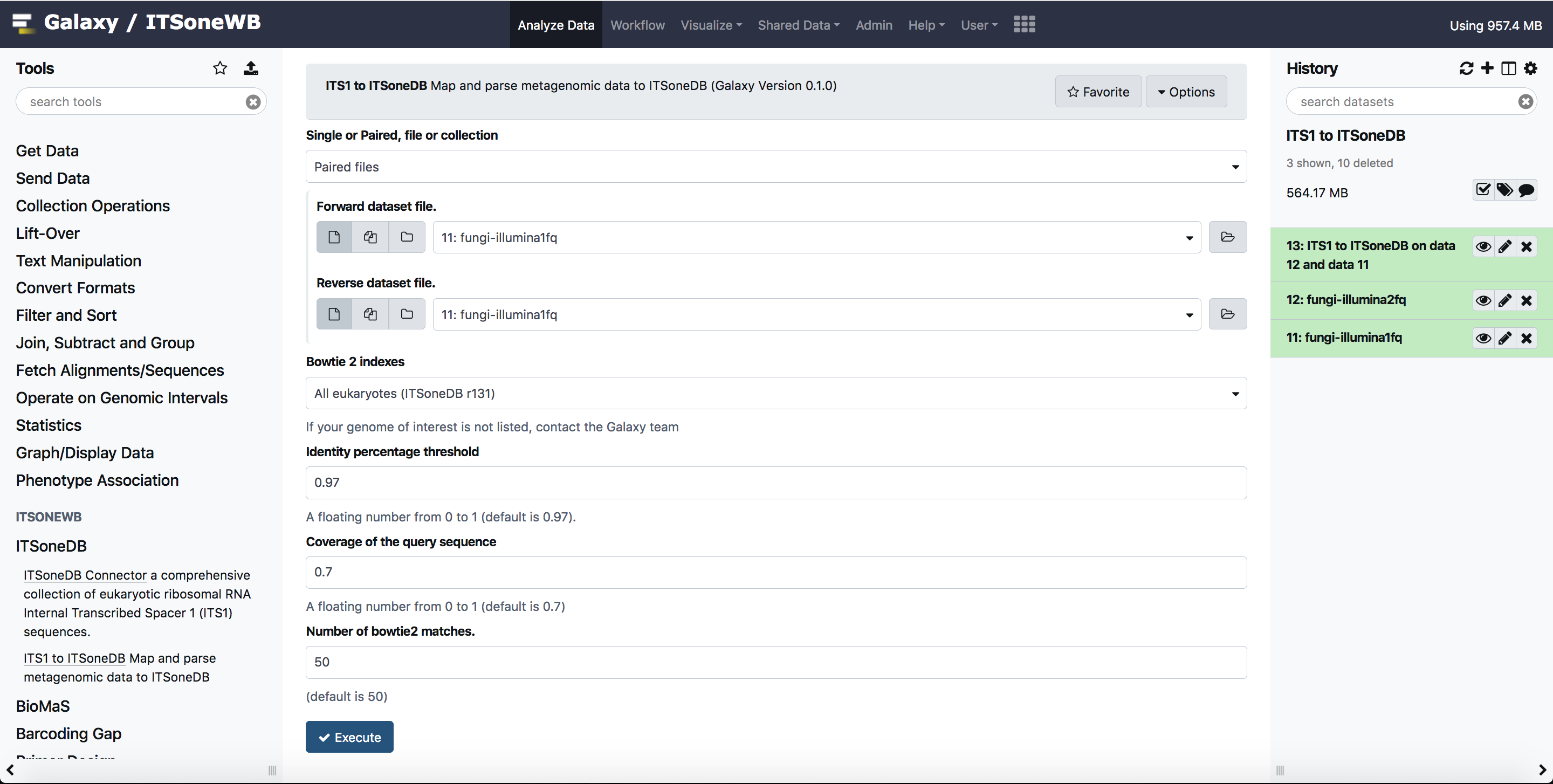
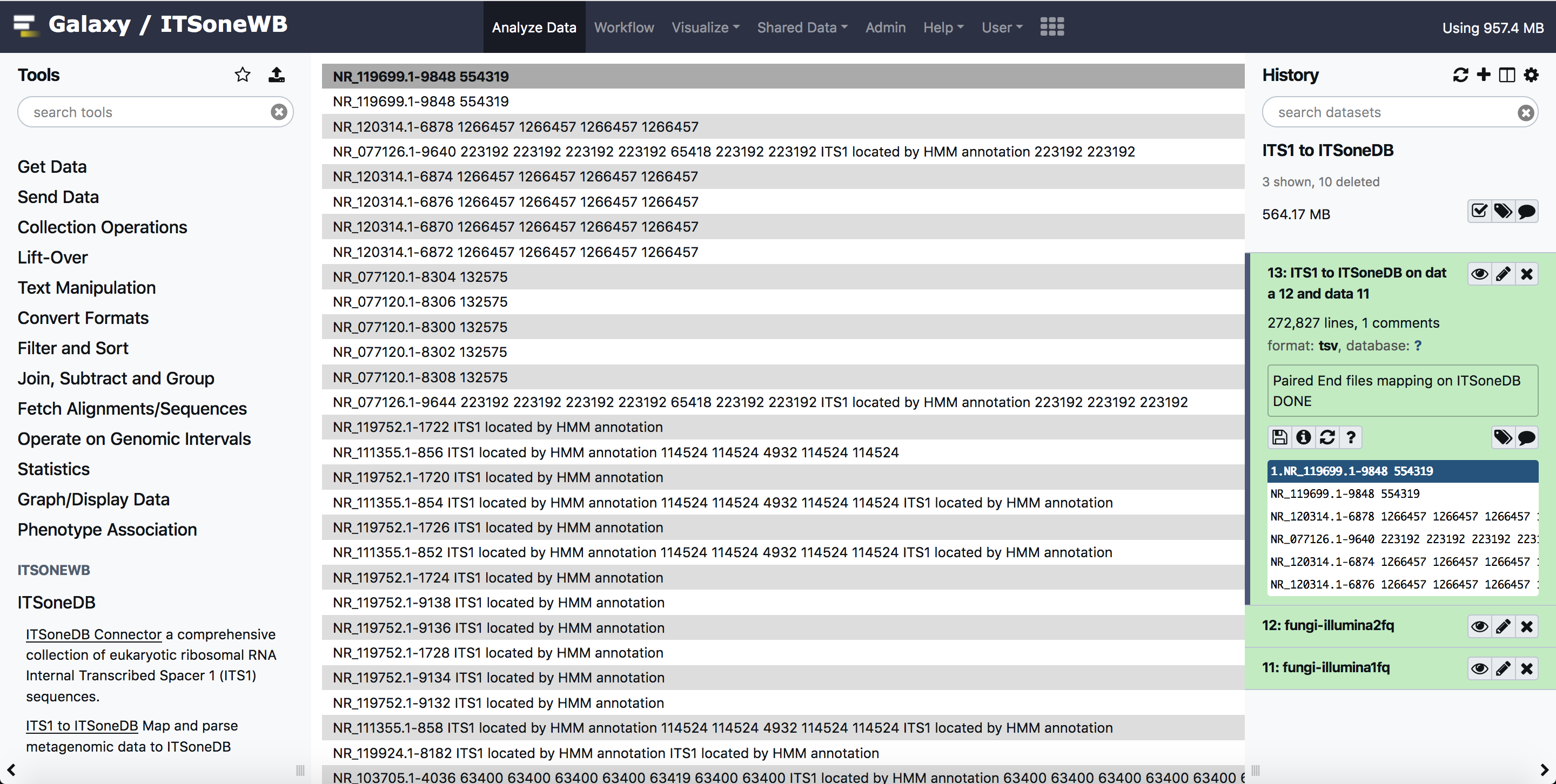
For testing purpose, we uploaded test input for BioMaS, in Galaxy shared library. The same inputs can be used for this tool. These can be exported to Galaxy history and used also by anonymous users.
Install on Galaxy¶
Galaxy is able to automatically solve conda dependecies when a tool is started.
To install the ITS1 to ITSoneDB tool on Galaxy:
Clone the ITSoneWb repository
git clone https://github.com/ibiom-cnr/itsonewb.git
Add the TSoneDB Connector entry in the galaxy
tool_conf.xmlfile with your favourite editor:<section name="ITSoneDB" id="itsonedb"> <tool file="/path_to_itsonewb/itsonewb/ITS1_parser_ITSoneDB_wrapper/ITS1_mapper_and_parser_ITSoneDB_wrapper.xml" /> </section>
Finally restart Galaxy.
Command line usage¶
Following the help page of the script:
# python ITS1_mapper_and_parser.py --help
usage: ITS1_mapper_and_parser.py [-h] [-f ITSONEDB_FASTA]
[-p1 [PAIRED1 [PAIRED1 ...]]]
[-p2 [PAIRED2 [PAIRED2 ...]]]
[-s [SINGLE [SINGLE ...]]]
[-i IDENTITY_PERCENTAGE] [-c COVERAGE]
[-b BOWTIE_INDEX] [-d OUTPUT_FOLDER]
[-t THREADS] [-n MATCH_NUMBER]
Map and parse metagenomic data to ITSoneDB
optional arguments:
-h, --help show this help message and exit
-f ITSONEDB_FASTA, --ITSoneDB_fasta ITSONEDB_FASTA
fasta file containing the ITSoneDB sequences
-p1 [PAIRED1 [PAIRED1 ...]], --paired1 [PAIRED1 [PAIRED1 ...]]
paired-end fastq file R1. Multiple files must be
listed space separated
-p2 [PAIRED2 [PAIRED2 ...]], --paired2 [PAIRED2 [PAIRED2 ...]]
paired-end fastq file R2. Multiple files must be
listed space separated
-s [SINGLE [SINGLE ...]], --single [SINGLE [SINGLE ...]]
single-end sam file
-i IDENTITY_PERCENTAGE, --identity_percentage IDENTITY_PERCENTAGE
identity percentage threshold (a floating number from
0 to 1, default is 0.97)
-c COVERAGE, --coverage COVERAGE
Coverage of the query sequence (a floating number from
0 to 1, default is 0.7)
-b BOWTIE_INDEX, --bowtie_index BOWTIE_INDEX
Path to the bowtie index folder
-d OUTPUT_FOLDER, --output_folder OUTPUT_FOLDER
Path to the folder where intermediate files will be
written
-t THREADS, --threads THREADS
number of threads
-n MATCH_NUMBER, --match_number MATCH_NUMBER
number of bowtie matches
An example of its application is available below:
python ITS1_mapper_and_parser.py -p1 example_files/fungi_R1_1.fq -p2 example_files/fungi_R1_2.fq -f itsonedb_biomas_indexes/ITS1_r131_plus_flanking_region.fna -b itsonedb_biomas_indexes/ITSoneDB_all_euk_r131 -d output
Paired End files mapping on ITSoneDB
DONE
The output file will be located in the output directory:
# cd output/
(its1-to-itsonedb) # ls
error.lst mapping_file.tsv paired_glocal.sam paired_local.sam
Install as standalone tool¶
The ITS1 to ITSoneDB mapper and parser tool can be found on the ITSoneWB Github repository.
Download the script:
wget https://raw.githubusercontent.com/ibiom-cnr/itsonewb/master/ITS1_parser_ITSoneDB_wrapper/ITS1_mapper_and_parser.py
The tool dependencies can be installed using conda, thorough its Bioconda channel:
conda create --name its1-to-itsonedb python=2.7 bowtie2=2.3.4.3 argcomplete=1.9.4 numpy=1.15.4 pysam tbb=2020.2 -c conda-forge -c bioconda
The command will create a new virtual environment called its1-to-itsonedb wich can be activated with:
conda activate its1-to-itsonedb
Bowtie indexes can be downloaded here.
Download and untar them:
wget http://cloud.recas.ba.infn.it:8080/v1/AUTH_3b4918e0a982493e8c3ebcc43586a2a8/ITSoneWB/itsonedb_r131_biomas_indexes.tar.gz
tar xvzf itsonedb_r131_biomas_indexes.tar.gz
Reference data¶
ITSoneDB (r131) BioMaS indexes can be downloaded here.
The archive includes the Bowtie2 indexes, i.e. bowtie2_indexes_rel131/ITSITSoneDB_all_euk_r131* files.
To include them in Galaxy, please refer to the Galaxy Project documnetation. The *loc files are on our github repository (ITS1_parser_ITSoneDB_wrapper/tool-data) with the corresponding tool_data_table_conf.xml entry.
Docker usage¶
The ITS1 to ITSoneDB mapper and parser tool is also packaged as Docker Container, hosted on DockerHub.
You can pull it from DockerHub with the following command:
docker pull ibiomcnr/its1-to-itsonedb
Note
The ITS1 to ITSoneDB docker container already includes the ITSoneDB reference data on /refdata/itsonedb_biomas_indexes/ITSoneDB_all_euk_r131.
The tool options are:
# ITS1-mapper-and-parser --help
usage: ITS1-mapper-and-parser [-h] [-f ITSONEDB_FASTA]
[-p1 [PAIRED1 [PAIRED1 ...]]]
[-p2 [PAIRED2 [PAIRED2 ...]]]
[-s [SINGLE [SINGLE ...]]]
[-i IDENTITY_PERCENTAGE] [-c COVERAGE]
[-b BOWTIE_INDEX] [-d OUTPUT_FOLDER]
[-t THREADS] [-n MATCH_NUMBER]
Map and parse metagenomic data to ITSoneDB
optional arguments:
-h, --help show this help message and exit
-f ITSONEDB_FASTA, --ITSoneDB_fasta ITSONEDB_FASTA
fasta file containing the ITSoneDB sequences
-p1 [PAIRED1 [PAIRED1 ...]], --paired1 [PAIRED1 [PAIRED1 ...]]
paired-end fastq file R1. Multiple files must be
listed space separated
-p2 [PAIRED2 [PAIRED2 ...]], --paired2 [PAIRED2 [PAIRED2 ...]]
paired-end fastq file R2. Multiple files must be
listed space separated
-s [SINGLE [SINGLE ...]], --single [SINGLE [SINGLE ...]]
single-end sam file
-i IDENTITY_PERCENTAGE, --identity_percentage IDENTITY_PERCENTAGE
identity percentage threshold (a floating number from
0 to 1, default is 0.97)
-c COVERAGE, --coverage COVERAGE
Coverage of the query sequence (a floating number from
0 to 1, default is 0.7)
-b BOWTIE_INDEX, --bowtie_index BOWTIE_INDEX
Path to the bowtie index folder
-d OUTPUT_FOLDER, --output_folder OUTPUT_FOLDER
Path to the folder where intermediate files will be
written
-t THREADS, --threads THREADS
number of threads
-n MATCH_NUMBER, --match_number MATCH_NUMBER
number of bowtie matches
Warning
Since the reference data are embedded in the Docker Container the path for the bowtie2 indexes is /refdata/itsonedb_biomas_indexes/ITSoneDB_all_euk_r131, to be used with -b option.
An example of its application is available below.
Create a directory to mount in the docker container. For example docker_test/its2itsonedb`. Then run the docker as follows:
# docker run -it -v $PWD/docker_test/its2itsonedb:/data ibiomcnr/its1-to-itsonedb ITS1-mapper-and-parser -p1 fungi_R1_1.fq -p2 fungi_R1_2.fq -f /refdata/itsonedb_biomas_indexes/ITS1_r131_plus_flanking_region.fna -b /refdata/itsonedb_biomas_indexes/ITSoneDB_all_euk_r131 -d .
Paired End files mapping on ITSoneDB
DONE
The output file will be located in the output directory, in this case $PWD/docker_test/its2itsonedb mounted in the docker container:
# cd docker_test/its2itsonedb
# ls
error.lst fungi_R1_1.fq fungi_R1_2.fq mapping_file.tsv paired_glocal.sam paired_local.sam